单细胞 RNA 测序 (single-cell RNA-seq, scRNA-seq) 技术作为最新一代测序技术,能够独立提供每个细胞的 RNA 表达谱,曾被Science杂志列为年度最值得关注的六大领域榜首。深入挖掘scRNA-seq数据信息对肿瘤研究、细胞免疫研究以及脑神经学研究等多个领域具有重要作用,对临床疾病的诊断以及治疗也有重要指导意义,因此scRNA-seq数据分析与建模是生物信息学的前沿和热点研究领域。随着测序技术的不断发展,规模更大的scRNA-seq数据不断涌现,发展准确高效的scRNA-seq数据分析与建模方法对生物医学研究具有重要意义。
近日,青岛科技大学数理学院人工智能与生物医学大数据团队于彬副教授,在生物信息学顶级期刊Briefings in Bioinformatics(IF=8.990) 上发表题为“scGMAI: a Gaussian mixture model for clustering single-cell RNA-Seq data based on deep autoencoder”的研究论文。报道了一种新的scRNA-seq数据聚类模型—scGMAI。该模型基于深度学习方法可以显著提高scRNA-seq数据的聚类结果并能准确识别细胞类型。于彬副教授为论文的第一作者及通讯作者,研究生陈晨为第二作者,青岛科技大学为第一完成单位。
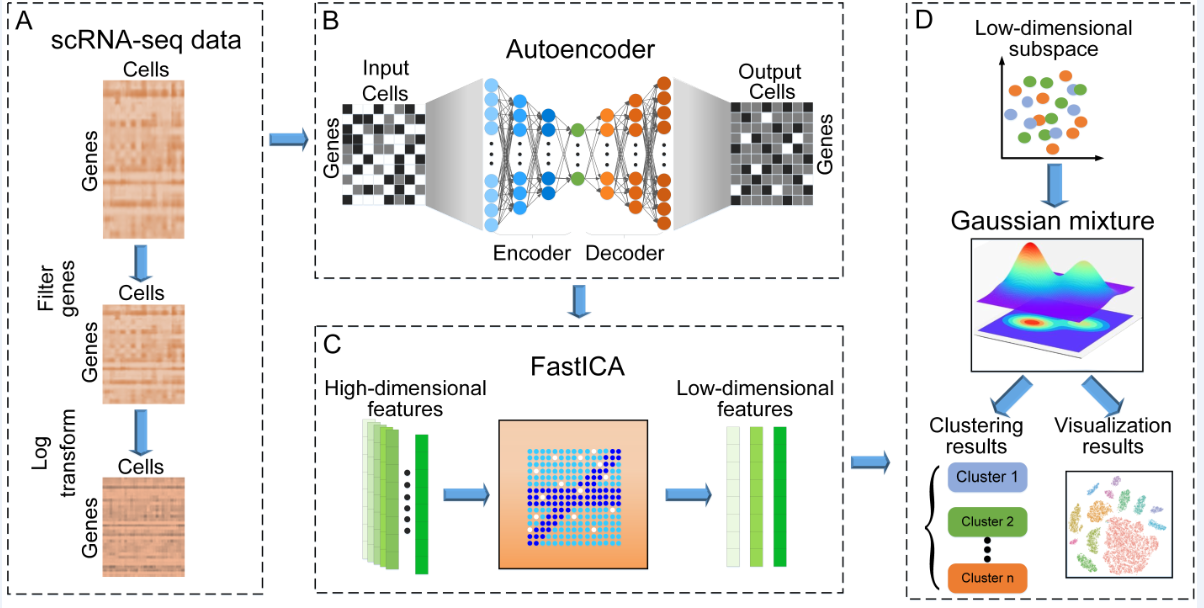
本研究首次使用深度自编码网络挖掘scRNA-seq数据中的重要信息并重构数据,提高了scRNA-seq数据下游分析能力。并且利用快速独立成分分析方法对数据降维,获取表征scRNA-seq数据的有效信息并提高计算效率。利用基于EM算法的高斯混合模型对细胞进行更为准确的聚类。在17个公开的scRNA-seq数据集上与其它先进聚类方法比较,结果表明scGMAI的性能明显优于其它聚类方法。scGMAI能够准确挖掘scRNA-seq数据中的基因表达信息并聚类细胞,为基因差异表达研究、细胞发育轨迹推断等下游分析提供有效的帮助,对临床疾病诊疗具有重要的指导意义。
文章链接:https://doi.org/10.1093/bib/bbaa316
Briefings in Bioinformatics是牛津大学出版社(Oxford Academic)出版的JCR一区顶级期刊,2020年的影响因子为8.990,在SCI收录的59个“Mathematical & Computational Biology”类期刊中排名第1,在SCI收录的79个“Biochemical Research Methods”类期刊中排名第3。
(撰稿:李磊; 审核:王明辉)




